INDICATIONS
MVASI® is a vascular endothelial growth factor inhibitor indicated for the treatment of:
MVASI®, in combination with intravenous fluorouracil-based chemotherapy, is indicated for the first- or second-line treatment of patients with metastatic colorectal cancer (mCRC).
MVASI®, in combination with fluoropyrimidine-irinotecan- or fluoropyrimidine-oxaliplatin-based chemotherapy, is indicated for the second-line treatment of patients with mCRC who have progressed on a first-line bevacizumab product-containing regimen.
Limitations of Use: MVASI® is not indicated for adjuvant treatment of colon cancer.
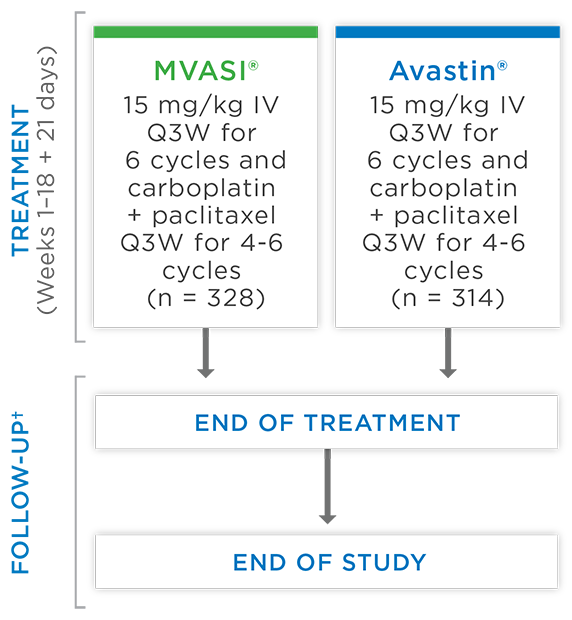
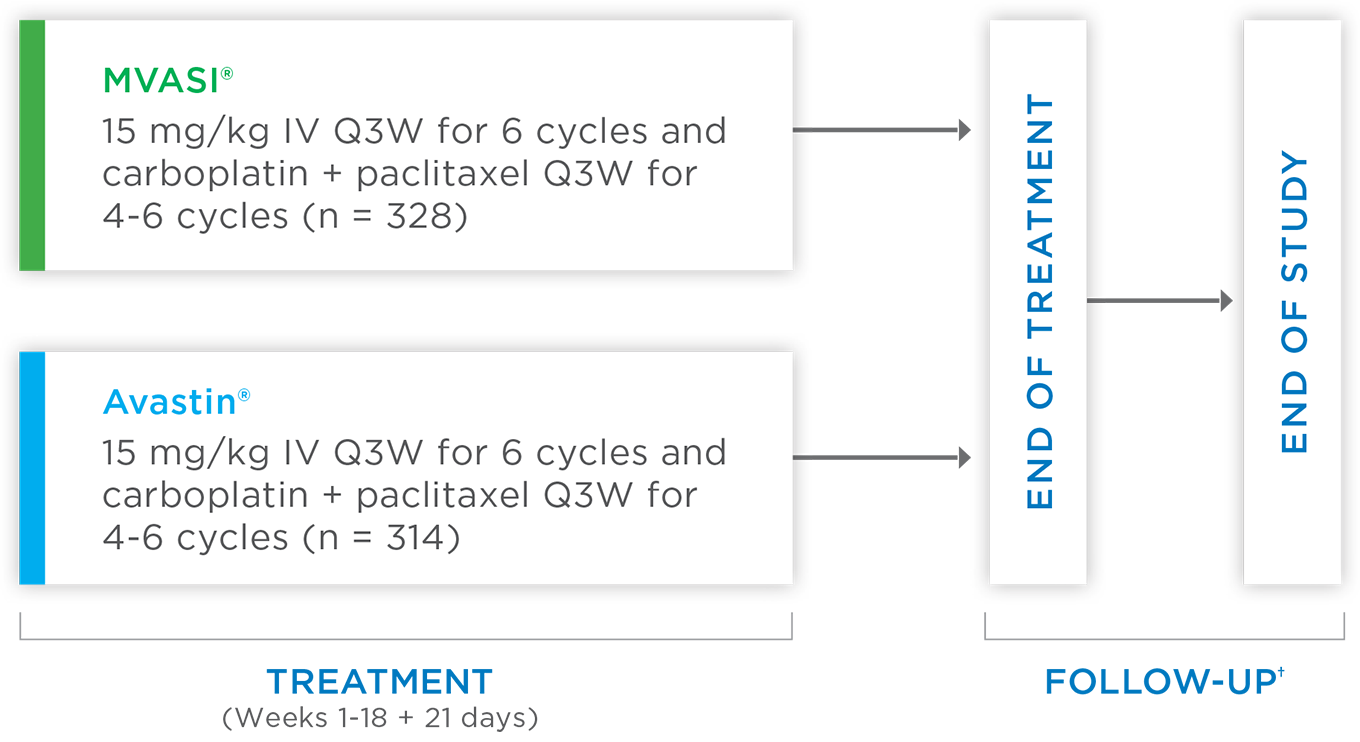
MVASI®, in combination with carboplatin and paclitaxel, is indicated for the first-line treatment of patients with unresectable, locally advanced, recurrent or metastatic non-squamous non-small cell lung cancer (NSCLC).
MVASI® is indicated for the treatment of recurrent glioblastoma (GBM) in adults.
MVASI®, in combination with interferon-alfa, is indicated for the treatment of metastatic renal cell carcinoma (mRCC).
MVASI®, in combination with paclitaxel and cisplatin or paclitaxel and topotecan, is indicated for the treatment of patients with persistent, recurrent, or metastatic cervical cancer (CC).
MVASI®, in combination with carboplatin and paclitaxel, followed by MVASI as a single agent, is indicated for the treatment of patients with stage III or IV epithelial ovarian, fallopian tube, or primary peritoneal cancer following initial surgical resection (OC).
MVASI®, in combination with paclitaxel, pegylated liposomal doxorubicin, or topotecan, is indicated for the treatment of patients with platinum-resistant recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer who received no more than 2 prior chemotherapy regimens (OC).
MVASI®, in combination with carboplatin and paclitaxel, or with carboplatin and gemcitabine, followed by MVASI as a single agent, is indicated for the treatment of patients with platinum-sensitive recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer (OC).
MVASI® is a trademark of Amgen, Inc.
Avastin® (bevacizumab) is a registered trademark of Genentech USA, Inc.